Bioinformatics Glossary
*** As I learn more, I will hopefully continue to add to this page for things that confused me or I thought would be helpful to compile together. As a warning, this is just a collection of notes and is not super organized.
Genetic Data
Reference Sequence Number
When I first started working on genetic data and especially with my PAX9 project, I was so confused by all of the different values that were associated with the gene:
Genomic reference: NG_013357.1 (GRCh 37)
Transcript reference: NM_006194.3
Ensembl gene ID: ENSG00000198807.8
Canonical transcript: ENST00000361487.6
GRCh 37
Before going into what all of these sequences are, first of all, what is GRCh37? I kept coming across this when I first tried to find data on my gene. GRCh37 stands for Genome Reference Consortium Human Build 37 and is generally synonymous with hg19. Similarly, hg38 can also be used to refer to the same genome build as GRCh38.
What is the difference between GRCh37 and GRCh38? GRCh38 was released after GRCh37 and has more updated annotations. Luckily, various tools such as from NCBI or Ensembl can be used for converting annotations.
NCBI Reference Sequence (RefSeq)
The accession number format:
NG_: gene/genomic region
NC_: plus (+) strand on chromosome
NT_: constructed genomic contigs (overlapping DNA fragments that are used to assemble the full sequence)
‘NC’, ‘NG’, ‘NT’ all refer to genomic sequences
NM_: mRNA, coding strand
NR_: non-coding RNA
NP_: protein
Also, if it begins with an ‘X’ liks ‘XP’ or ‘XM’, it probably means that the sequence is from computational prediction.
What about CDS? That is the coding region of a gene that is translated to protein. Therefore, it excludes the UTR and introns.
Ensemble
ENST: transcipt
ENSG: gene (One gene can have many corresponding transcripts.)
ENSP: protein
SNPs
dbSNP: Single Nucleotide Polymorphism Database from NCBI, used for SNPs
rs: RefSNP, also used to refer to a specific SNP
Types of Files
FASTA
FASTA is a type of text-based file that is used to store a nucleotide or amino acid sequence. In R, there is the read.fasta() function from the ‘seqinr’ package. It’s written in the following format:
>3LWB:A|PDBID|CHAIN|SEQUENCE MSANDRRDRRVRVAVVFGGRSNEHAISCVSAGSILRNLDSRRFDVIAVGITPAGSWVLTDANPDALTITNRELPQVKSGS GTELALPADPRRGGQLVSLPPGAGEVLESVDVVFPVLHGPYGEDGTIQGLLELAGVPYVGAGVLASAVGMDKEFTKKLLA ADGLPVGAYAVLRPPRSTLHRQECERLGLPVFVKPARGGSSIGVSRVSSWDQLPAAVARARRHDPKVIVEAAISGRELEC GVLEMPDGTLEASTLGEIRVAGVRGREDSFYDFATKYLDDAAELDVPAKVDDQVAEAIRQLAIRAFAAIDCRGLARVDFF LTDDGPVINEINTMPGFTTISMYPRMWAASGVDYPTLLATMIETTLARGVGLH >3LWB:B|PDBID|CHAIN|SEQUENCE MSANDRRDRRVRVAVVFGGRSNEHAISCVSAGSILRNLDSRRFDVIAVGITPAGSWVLTDANPDALTITNRELPQVKSGS GTELALPADPRRGGQLVSLPPGAGEVLESVDVVFPVLHGPYGEDGTIQGLLELAGVPYVGAGVLASAVGMDKEFTKKLLA ADGLPVGAYAVLRPPRSTLHRQECERLGLPVFVKPARGGSSIGVSRVSSWDQLPAAVARARRHDPKVIVEAAISGRELEC GVLEMPDGTLEASTLGEIRVAGVRGREDSFYDFATKYLDDAAELDVPAKVDDQVAEAIRQLAIRAFAAIDCRGLARVDFF LTDDGPVINEINTMPGFTTISMYPRMWAASGVDYPTLLATMIETTLARGVGLH
The first line is used to describe the folowing sequence and it is indicated by the “>” sign. As you can see above, multiple sequences can be found in one FASTA file.
Fastq
Similar to FASTA, except from DNA sequencing (commonly for illumina). It also includes the PHRED score which is a measure of the quality of the reading for each base. It is denoted by a single ASCII character.
@ SequenceID
CATGGGCAGCCGAGAGATTGCGA
+
K<=gux;YZ[bcs3^_`a;<|}
The first line contains the description like fasta but is indicated by the “@” sign instead. After which, the nucleotide sequence is shown on the 2nd line. The third line starts with a “+” sign. Finally, the 4th line contains the quality scores.
SAM/BAM files
SAM and BAM files contain the same information, the difference is that BAM files are binary versions of a SAM file. SAM stands for “Sequence Alignment/Map” and contain information about sequences aligned to a reference sequence, and is generated by next generation sequencing.
SDF (structure data file)
** MOL files follow the same format but are only for a single molecule while SDF files can contain multiple
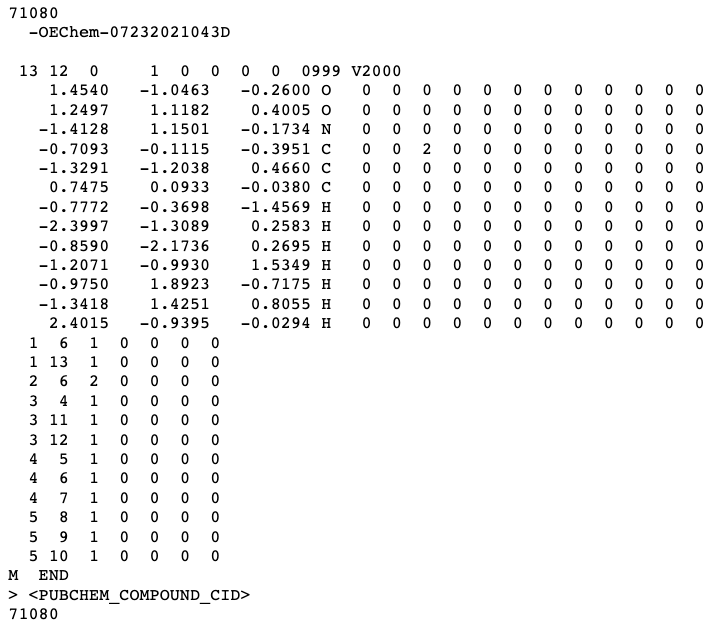
Three line header:
Name of Molecule (71080)
Software to generate the file
Comment (blank above)
*** Even if lines are left blank. Still need the lines there for file to function properly.
Counts line
Number of atoms: 13
Number of bonds: 12
Atoms (one line for each atom)
X coordinate: 1.450
Y coordinate: -1.0463
Z coordinate: -0.2600
Atom symbol: O (oxygen)
Bonds (one line for bond)
Index of atom 1
Index of atom 2
Type of bond (1=single, 2=double, 3=triple)
M END : Required at end.
Metadata (data that provides info on other data)
Starts with header that begins with >
Name of data field is written in <>.
E.g. > <PUBCHEM_COMPOUND_CID>
Multiple molecules are separated with four dollar signs: $$$$.
VCF
VCF stands for variant call format and stores different variants of a gene.
Bioinformatic Resources/Tools
BLAST
BLAST stands for Basic Local Alignment Search Tool. It finds areas of similarity between sequences of nucleotides or amino acids using a scoring matrix. There are many types of blast including blastn, blastp, blastx, tblastn, etc.
Query cover: how much of the query sequence actually overlapps with the aligned sequence.
E value (expected value): smaller is better (significant). It basically means how many hits would you get just randomly (based on the quality of score and the length of the query)
Percent Identity: percentage of characters matching in the query and aligned sequence
BRENDA
BRENDA contains enzyme information such as the reaction diagram, links to research papers, KM values, IC50, inhibitors for the enzyme, etc.
ExPASy
ExPASy or Expert Protein Anaylsis System can be used to find the molecular weight of a sequence of amino acids, theoretical pI, instability, extinction coefficient (for spectrophotometry), etc.
PDB
PDB or Protein Data Bank contains 3D files of mostly proteins. For each protein, it tells you the associated research paper, number of chains, any ligands present already in the structure, resolution, etc.
Rosalind
Different from the other resources on this list, this is a great tool for practicing bioinformatics coding. Here’s a link to my profile to see my progress so far.
Micellaneous
BLOSUM (Block Storing Matrix) and PAM
Both used as a measure of similarity between proteins. PAM is for closely related while BLOSUM is for distantly related. For BLOSUM, BLOSUM45 would be used for proteins more distant from each other than BLOSUM90.
E.C. Number
You can categorize enzymes based on their EC number (Enzyme Committee number) based on the reaction they catalyze. For example, D-alanine D-alanine ligase has an E.C. number of 6.3.2.4. The “6” indicates that it is a ligase (joining two molecules using ATP).
Hamming Distance
What is the minimum number of nucleotides that need to be changed for one string to transform into another (if they are the same length). In biology, one application is in identification: in illumina sequencing, you can sequence multiple samples together and they can be differentiated due to adaptor sequences which have barcode/index sequences that are unique to each sample.
| Sequence 1 | Sequence 2 (Hamming dist=1) | Sequence 3 (Hamming dist=2) | Sequence 3 (Hamming dist=2) | |
|---|---|---|---|---|
| Original | ACCG | ACTG | ACTT | GCTT |
| 1 Mutation | “did not change” | ACCG | ACCT | GCCT |
| 2 Mutations | “did not change” | - | ACCG | GCCT |
| 3 Mutations | “did not change” | - | - | ACCG |
However, if one mutation occurs per sequence, than you need at least a hamming distance of 2(error)+1. For example: if you’re starting sequences are “ACCG” and “GCTT”, what if you get the sequence “ACCT”? Is that the sequence “ACCG”–> “ACCT”? Or “GCCT”–> “ACCT”? Therefore, if you have one error per sequence, you need a hamming distance of atleast 3 .
K-mers
Subsequences of length k of a nucleotide sequence.
def findkmers(k, seq):
dict1={}
a = 0
b= k
k_mer= seq[a:b]
while len(k_mer)==k:
if k_mer in dict1:
dict1[k_mer]+= 1
else:
dict1[k_mer]= 1
a+=1
b+=1
k_mer= seq[a:b]
print (dict1)
def main():
my_seq1 = "CAGCCCAATC"
print("For the sequence: ", my_seq1)
for i in range (1,5):
print("The", str(i)+"-mers are :")
findkmers(i, my_seq1)
main()## For the sequence: CAGCCCAATC
## The 1-mers are :
## {'C': 5, 'A': 3, 'G': 1, 'T': 1}
## The 2-mers are :
## {'CA': 2, 'AG': 1, 'GC': 1, 'CC': 2, 'AA': 1, 'AT': 1, 'TC': 1}
## The 3-mers are :
## {'CAG': 1, 'AGC': 1, 'GCC': 1, 'CCC': 1, 'CCA': 1, 'CAA': 1, 'AAT': 1, 'ATC': 1}
## The 4-mers are :
## {'CAGC': 1, 'AGCC': 1, 'GCCC': 1, 'CCCA': 1, 'CCAA': 1, 'CAAT': 1, 'AATC': 1}Needleman-Wunsch algorithm
Global alignment
ORF
ORF stands for open reading frame. When reading in a DNA strand, it’s important to note that either strand can be the coding strand! Therefore, there are 6 reading frames per DNA strand (not 3 like I previously would have immediately thought). You do however need to reverse complement the given DNA strand.
Smith-Waterman algorithm
Local alignment
References
http://useast.ensembl.org/info/website/tutorials/grch37.html
https://bitesizebio.com/38335/get-to-know-your-reference-genome-grch37-vs-grch38/
https://www.ncbi.nlm.nih.gov/books/NBK50679/#RefSeqFAQ.what_is_a_reference_sequence_r
Kerfeld and Scott, PLoS Biology 2011
https://ryanstutorials.net/bash-scripting-tutorial/bash-script.php
http://www.nonlinear.com/progenesis/sdf-studio/v0.9/faq/sdf-file-format-guidance.aspx
Cara Zou
Interests include dentistry and computer science.